Growing up With Cystic Fibrosis
Original Editor - Darragh Barron, Tara Campbell, Kevin Custodio, Kourosh Foroughi, Ashlea Masters, Kirstin Moynihan and Laura Wickham as part of the Queen Margaret University's Current and Emerging Roles in Physiotherapy Practice Project
Top Contributors - Laura Midori Wickham, Kim Jackson, Vidya Acharya, 127.0.0.1, Rachael Lowe, Michelle Lee, Naomi O'Reilly, WikiSysop and Adam Vallely Farrell
Introduction[edit | edit source]
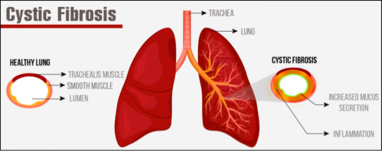
Cystic Fibrosis is an inherited disease of the mucus and sweat glands (exocrine glands) affecting mostly the lungs, liver, pancreas and intestines. It causes damage to lung tissue, inflammation, and acute susceptibility to bacterial infections. There is an abnormal gene, called Cystic Fibrosis Transmembrane Conductance Regulator (CFTR), which results in the production of thick, sticky mucus which blocks the airways in the lungs resulting in frequent lung infection. The thick mucus also blocks the ducts in the pancreas which in turn blocks digestive pancreatic enzymes from reaching the small intestine and performing their normal function. For more information on the pathophysiology see Cystic Fibrosis.
Growing Up with Cystic Fibrosis[edit | edit source]
Childhood[edit | edit source]
School can be a huge source of anxiety and depression build up among children living with cystic fibrosis.[2] Appointments, such as physiotherapy, is time-consuming, sometimes at the expense of a child’s social life, adding to stress and anxiety. Positively, children with cystic fibrosis often find supportive friends who help with care.[3] Children with cystic fibrosis may be teased or picked on at school for reasons such as but not limited to:
- Being underweight and small for their age (some people with cystic fibrosis experience a delayed onset of puberty, which may cause anxiety or insecurity.)
- Having a persistent cough
- Taking tablets and capsules with meals
- Eating a different diet from classmates
- Missing school for treatments[4][5]
Teenage Years[edit | edit source]
During teenage years there is a chance that children may neglect their physiotherapy and diet, further decreasing mood, attitude and adherence. Teenagers may need sympathetic treatment and counselling to help them deal with some of these issues. Cystic fibrosis requires a level of special involvement from teachers, which could include discussions with parents or even practical help. The most serious psychological problems of cystic fibrosis occur in adolescence when the rebellious behaviour shown by most teenagers may pose a threat to the health of someone with cystic fibrosis. Parents of children living with cystic fibrosis may be anxious about how their child will cope with school. Teachers can provide invaluable reassurance by making a special effort to meet parents before the child comes into their class. Staff at schools can prove invaluable when a child with cystic fibrosis changes a class or teacher. Teachers may find that brothers or sisters of children with cystic fibrosis have problems at school too.[6]These issues as well as the unpredictable outcome of cystic fibrosis may be very stressful for teenagers, requiring sympathetic understanding and counselling.
Psychosocial Factors[edit | edit source]
Psychosocial environment in early life is very important on the future health of all young children. For example, children who move frequently in early life are at increased risk of inferior physical health,[4] behavioural problems and lower educational achievement. Family structure and function can directly influence anxiety and depression levels in a child with cystic fibrosis. Family functioning can be measured by family stress, parental capacity/mental health, family coping and resilience (including adjustment to diagnosis) and the one on one relationship between caregiver and the person living with disease. Elements of family functioning and social demographic factors are well known to have a major impact on normal child development and therefore are also likely to have significant influences on the progression and severity of cystic fibrosis.[4][7][7][4][8][9] Health professionals working with people with cystic fibrosis report significant improvements in children’s health outcomes following positive changes to the family or social environment.[10] Conversely, the impact of chronic disease on the psychosocial and social economic status of individuals or families can be considerable.[7] Additionally, pulmonary exacerbations may worsen depression and anxiety, from the impact of demoralisation, stress and inflammation.[6]
The transition from childcare to an adult cystic fibrosis service comes with a lot of changes[11]. People living with cystic fibrosis sometimes experience daily treatment, physical restrictions, psychosocial morbidity and health decline. Some people living with cystic fibrosis may find that coping with the disease elevates levels of stress, anxiety and depression[12]. Studies have found that individuals with cystic fibrosis have higher rates of both depression and anxiety compared to the general population.[4][8][9][10][7][13] Moreover, psychological symptoms in both people living with cystic fibrosis and parents have been associated with decreased lung function, lower body mass, worse adherence, worse health-related quality of life, more frequent hospitalisations and increased healthcare costs.[4][3] In addition, some family and social variables change throughout this time period and also influence disease progression.[10] Studies have shown that people with cystic fibrosis, as well as parents who take care of children with cystic fibrosis, are more likely to experience anxiety than people in the general population. Given these high rates of depression and anxiety and their effects on quality of life and key health outcomes, the Cystic Fibrosis Foundation and the European Cystic Fibrosis Society supported the formation of an International Committee on Mental Health in CF (ICMH). The ICMH provides guidelines and recommendations for treatment and screening for depression and anxiety.
Anxiety and Cystic Fibrosis[edit | edit source]

Anxiety is a normal emotion that comes and goes in response to fears or worries about changes in health, work, relationships or money.[14] A person may have an anxiety disorder if the anxiety does not go away, gets worse over time and prevents them from participating in ordinary daily activities. An anxiety disorder is different than normal anxiety in that it can occur for an extended period of time and interfere with the ability to manage the symptoms of cystic fibrosis effectively which may impact on quality of life. Some people living with cystic fibrosis also experience a very specific form of anxiety centered on medical procedures. Anxiety is one of the most common emotional issues that people face. People living with cystic fibrosis or who have a child with cystic fibrosis may experience a great deal of stress. Making time for daily treatments, remembering to take medications, missing out on life experiences as a result of being hospitalised for an infection all cause stress and anxiety, which affect emotional wellness.
Anxiety disorder affects both physical and emotional health, for example, some people with untreated anxiety:
- Are less likely to manage their treatment plans
- Have a lower body mass index (BMI)
- Tend to have worse lung function
- Experience more hospitalizations
- Often have higher health care costs
- Experience a lower quality of life[3]
Symptoms of Anxiety include
- Worrying too much
- Exaggerated worry
- Restlessness
- Irritability
- Muscle tension
- Headaches
- Sweating
- Difficulty concentrating
- Trouble falling asleep or staying asleep
- Fatigue
- Trembling
- Startling easily[3]
Children and teenagers may have additional symptoms, including but not limited to worries about:
- School or sports performances
- Being on time
- Natural and man-made disasters, such as war or earthquakes
- Fitting in with peers
- Performing homework for long periods of time
- Redoing homework assignments
- Being perfectionists
- Getting approval
- Getting reassurance[3]
Anxiety About Medical Procedures[edit | edit source]
For people living with cystic fibrosis, ‘procedural anxiety’ is particularly most important. Procedural anxiety is excessive fear of a medical or surgical procedure that results in serious stress or avoidance. Patients may experience anxiety in anticipation of or during procedures. Avoidance due to procedural anxiety can have negative health consequences.[5] Many people with cystic fibrosis need to have invasive medical procedures, such as placement of feeding tubes. Feeling nervous about medical procedures is natural but the exaggerated fear or phobia that some people with cystic fibrosis experience before medical procedures is not normal when it interferes with their ability to manage their cystic fibrosis effectively.[3]
Identifying Anxiety[edit | edit source]
Screening using outcome measures, usually in the form of a short survey, can often identify feelings of anxiety, such as nervousness, uncontrollable worry or difficulty performing daily activities, such as going to work or taking care of things at home. If the survey results suggest the presence of anxiety disorder, your cystic fibrosis care team further evaluation and treatment may be necessary. Anxiety can be treated successfully, but only if the symptoms are properly[4][3] identified.
Some examples of outcome measures are:
1. Patient Health Questionnaire 9 (PHQ-9) - Includes an item to assess suicide risk.
2. Generalised Anxiety Disorder 7-item (GAD-7) - A scale for annual screening of adolescents (ages 12 years and older) and adults with cystic fibrosis and offered annually to at least one primary caregiver of children with cystic fibrosis (ages 0–17 years).
PHQ-9 and GAD-7 are free, brief, reliable and valid. Both provide optimal cut-off scores for detecting psychological symptoms and are available in all major languages.
For procedures on screening and treatment of depression and anxiety for individuals with CF (ages 12–adulthood) please see flow chart below[6]

Treating Anxiety[edit | edit source]
Cognitive Behavioural Therapy[edit | edit source]
Cognitive behavioural therapy can help to identify and change unrealistic or unhealthy thoughts, emotions and behaviours. After identifying unhealthy thoughts, emotions and behaviours, the challenge is to replace them with more effective thoughts and behaviours. Learning relaxation techniques and deep breathing can also be a useful tool in managing anxiety.[4][3]
Medication[edit | edit source]
Medication can help restore the balance of brain chemicals and is typically prescribed by a psychiatrist who is a medical doctor with special training in identifying and treating anxiety. Although these medications are commonly known as antidepressants, they also are very effective at helping people with anxiety. One class of antidepressant medication commonly prescribed to treat anxiety is serotonin reuptake inhibitors (SSRIs). SSRIs work by preventing the reabsorption of the chemical serotonin, which can relieve anxiety. These medications can begin working within one to two weeks, but their full effects might not be experienced for two to three months. For people with more severe anxiety or anxiety that does not improve with either talk therapy or medication, treatment may be a combination of the two.[4][3]
Treatment of Anxiety Related to Medical Procedures[edit | edit source]
Treatment of anxiety, such as a phobia related to medical procedures, begins with cognitive behavioural therapy. If anxiety levels before procedures do not improve, medications called benzodiazepines may be prescribed beforehand. Benzodiazepines are sedatives that help a person relax. They are for short-term use only, because they can be habit-forming.[4][3][2] Offering advice on effective self-help measures is an important to step to empower the patient. Advice such as:
- Being physically active. Exercise can help reduce stress.
- Practicing relaxation techniques.
- Avoiding alcohol or drugs.
- Avoiding caffeine and cigarettes, which can increase anxiety levels.
- Practicing good sleep habits. Getting enough sleep and going to bed and waking up on a consistent schedule.
- Getting outside or in nature for 30 minutes each day.
- Making time for hobbies.
- Continuing with their treatment plan.
- Joining an anxiety support group and about talking about their problems with people who have the same experience can help to feel less alone.
Although these activities are not a substitute for professional care, they can make a real difference to anxiety levels.
Depression in Cystic Fibrosis[edit | edit source]

People living with cystic fibrosis can be depressive in mood or lose the interest in most activities.[2] Unlike ordinary sadness, clinical depression can last for a long time if not treated. People who have depression can have extended periods where they feel hopeless and lose interest in things they normally would enjoy. Studies measuring psychological distress in individuals with cystic fibrosis have found elevated rates of both depression and anxiety. The prevalence of depression ranges from 8% to 29% among children and adolescents, and 13–33% among adults, anxiety in adults has ranged from 30% to 33%.[5][4] Researchers found that people with cystic fibrosis and parents who take care of children with cystic fibrosis are more likely to experience depression than people in the general population.[4]Caregivers have also been reported to have elevations in depression scores ranging from 20% to 35%.[8][9] Evidence indicates that when a parent reported elevated depressive or anxious symptoms, the adolescent with cystic fibrosis was more than twice as likely to also experience depression and anxiety. Untreated depression can affect both physical and emotional health, and interfere with the ability to take care of theselves. Risk for suicide is a core component of depression, is a major cause of death among adolescents and adults in the general population and in recent years found to be about 1.6% of all deaths among people living with cystic fibrosis.[10] People with untreated depression:
- Are less likely to manage their treatment plans
- Tend to have worse lung function
- Have a lower body mass index (BMI)
- Experience more hospitalisations
- Often have higher health care costs
- Experience a lower quality of life[10]
Symptoms of Depression[edit | edit source]
Depression may be present if five or more of the following symptoms are experienced for two weeks or more:
- Sadness
- Loss of energy*
- Feelings of hopelessness or worthlessness
- Loss of enjoyment in things you or your child once liked
- Problems concentrating
- Uncontrollable crying
- Problems making decisions
- Irritability
- Sleeping more than usual
- Trouble falling asleep or staying asleep
- Unexplained aches and pains
- Stomach aches or other digestive problems
- Loss of interest in sex
- Sexual problems
- Headaches
- Loss of appetite and weight loss*
- Weight gain
- Thoughts of suicide
- Suicide attempts
- Some symptoms of depression, such as fatigue or weight loss, also can be symptoms of cystic fibrosis.[10]
What Increases the Risk for Depression?[edit | edit source]
• Physical and emotional abuse.
• Certain medications.
• Family history.
• Personal conflicts or arguments.
• Death or another emotional loss.
• Significant life events, even positive ones.
• Other personal issues.
• Substance abuse. Almost 30 percent of people with depression abuse alcohol or drugs.[10]
Identifying Depression[edit | edit source]
Screening using outcome measures, usually in the form of a short survey, can often identify feelings of depression, such as nervousness, uncontrollable worry or difficulty performing usual activities, such as going to work or taking care of things at home. f the survey results suggest that you or your child may have depression, your cystic fibrosis care team may recommend further evaluation and treatment if necessary. Some examples of outcome measures are:
1. Patient Health Questionnaire 9 (PHQ-9) - Includes an item to assess suicide risk.
2. Generalised Anxiety Disorder 7-item (GAD-7) - A scale for annual screening of adolescents (ages 12 years and older) and adults with cystic fibrosis and offered annually to at least one primary caregiver of children with cystic fibrosis (ages 0–17 years).
PHQ-9 and GAD-7 are free, brief, reliable and valid. Both provide optimal cut-off scores for detecting psychological symptoms and are available in all major languages.[6]
Treating Depression[edit | edit source]
Talk Therapy Talk therapy involves meeting with a health care professional who specializes in treating depression, discusses your issues and works with you to develop solutions. Common types of talk therapy for treating depression include:
- Cognitive Behavioral Therapy can help you identify and change unrealistic or unhealthy thoughts, emotions and behaviors. After identifying unhealthy thoughts, emotions and behaviors, you challenge and replace them with more effective thoughts and behaviors.
- Interpersonal Therapy can help you identify issues that may be causing problems for you, such as conflicts in your relationships or unresolved grief. Specific preventive strategies may be developed to reduce the risk of anxiety and depression in cystic fibrosis. For example, training in specific problem-solving and cognitive behavioural skills can decrease anxiety and improve resilience.[10][7]
- Medication is typically prescribed by a psychiatrist who is a medical doctor with special training in identifying and treating mental health conditions. Medication can help to restore the balance of brain chemicals, which are called neurotransmitters. Neurotransmitters, such as dopamine and serotonin, are chemicals that relay signals between nerve cells. When they are out of balance, they can negatively affect your mood. A common class of antidepressant medication is serotonin reuptake inhibitors (SSRIs). SSRIs work by preventing the reabsorption of serotonin, which can relieve depression. Antidepressant medications begin working within one to two weeks, but you might not experience their full effects for two to three months. If you have not started to feel better after several weeks, tell your doctor. He or she can adjust your medication dosage or prescribe different medications to provide the best effect. If you have not begun to feel better after several weeks, tell your doctor or care team.[4] For people with more severe depression or depression that does not improve with either talk therapy or medication, treatment may be a combination of the two.[4]
Physiotherapy Management[edit | edit source]
As CF is a longterm condition it is important to include education and self care as part of the treatment programme. Many treatment strategies are appropriate for all ages but some may require modifications for younger individuals such as infants or toddlers. Many of the physiotherapy interventions will focus on keeping the airways clear but exercise also plays a vital role in the management of this condition. Airway Clearance Techniques (ACTs) are done to help individuals breath easier and stay healthy by loosening thick and sticky lung mucus allowing the individual to clear them by coughing. By mobilizing and getting rid of this mucus it reduces the chances of lung infection and it also improves lung function.[13]
Active Cycle of Breathing Technique[edit | edit source]
Active Cycle of Breathing Technique (ACBT) consists of breathing exercises that are used to mobilize and clear excess airway secretions and mucus.[13] Studies have shown that ACBT can be used as an effective and efficient technique for mobilizing and clearing secretions in persons with cystic fibrosis.[13]
- ACBT can be introduced to children with CF as “huffing games” by the age of about 2 years old
- Children can gradually start taking more responsibility for practicing this cycle of breathing around the ages of 8 or 9 allowing them to become a bit more independent
- It can be done in any position according to the requirements of each person. Most individuals prefer the sitting position as it is effective.
There are 3 components to ACBT:
- Breathing control (BC) - Relaxed breathing at the individual's normal rate and depth. The person is encouraged to “tummy breath” instead of doing deep chest breathing.
- Thoracic Expansion Exercises (TEE) - These breaths emphasize the breath going in. The person breaths in deeply and usually holds their breath at the end when they are full of air for 3 seconds and then lets their breath out in a relaxed and unforced manner. This works to get the secretions unstuck and moving. The 3 second hold is important to allow the air to reach into the obstructed regions that it might not normally get to during relaxed breathing due to the build up of mucus in those areas.
- Forced expiratory technique “Huff” (FET) - Combination of 1 or 2 “huffs” and periods of breathing control afterwards. A “huff” is done by opening your mouth and your throat and pushing air outwards as if the person is trying to fog up a mirror. Huffing works to loosen and mobilize excess bronchial secretions from the smaller airways towards the larger more central airways that are closer to the throat and mouth. When the secretions reach these central airways, a huff can stimulate a cough which can bring the secretions and mucus to the mouth allowing the individual to spit them up into a tissue or swallow them into the stomach.[13]
ACBT is repeated until the huff becomes dry sounding and non-productive or the individual needs a rest. The total treatment time is usually between 10 and 30 minutes.
The order of the breathing exercises, the position of the individual and the length of time and number of treatments in a day will be determined and change depending on the day and whether the person is clinically stable or is going through an acute exacerbation of pulmonary infection at the time.[13]
Autogenic Drainage[edit | edit source]
Autogenic Drainage is an airway clearance technique used to avoid airway closure that may be caused by coughing and manoeuvering. It allows the individual to reach the highest possible airflow in different generations of bronchi by controlled breathing. The technique is based on physics, fluid dynamics, lung anatomy, respiratory physiology and breathing mechanics.
Airflow causes shearing forces that help to mobilize secretions that have built up on the airways. It is key to monitor the breaths going in and the breaths going out in order to create the necessary shearing forces in the right areas to clear the airways.
- During the breath in, the speed of the breath must not be too quick as to avoid moving secretions further into the lungs
During the breath out, the optimal speed of airflow out must be matched with correct depth of the secretions within the lungs. To localize the secretions there are 3 feedback signals the person and anyone assisting with this technique should be aware of:
- Sound of the secretions
- Feeling within the chest
- Proprioceptive feedback (Proprioception refers to the unconscious perception of movement and spatial orientation arising from stimuli within the body itself. These stimuli are detected by nerves within the body as well as canals in the ears.[13]
For infants and non-cooperative individuals who suffer from cystic fibrosis Assisted Autogenic Drainage (AAD) can be used as an alternative. It is performed in a gentle and progressive way, using the person’s natural breathing pattern and stabilizing the infants abdominal wall. A gentle increase in pressure on the chest during the person’s breath is done to guide the breathing towards the desired lung volume. The hands gradually restrict the breath being taken in to stimulate the persons to breath out slightly more than the previous cycle. Feedback by feeling or hearing the secretions move.[13] There a few precautions that need to be considered when performing this technique:
- Special care should be taken of spastic or swollen airways in all airway clearance techniques. Secretions can be made easier to mobilize by means of pharmaceutical management.
- No excessive force is needed. This could lead to resistive responses by the individual.
- Patience is essential in this technique.
- The individual should be sitting upright and be supported to avoid a slumped sitting position which in turn may lead to some gastro-oesophageal (heart burn) reflex during treatment.[13]
Postural Drainage[edit | edit source]
Postural drainage uses various body positions to allow gravity to assist in draining mucus from the outer airways of the lungs towards the central airways so that they can be cleared by a cough. Today modified postural drainage positions are the accepted method of treatment for children and adults with cystic fibrosis. Some of these modifications include eliminating the head down position as it increases the changes for gastroesophageal reflux, can be quite uncomfortable and has been shown to decrease oxygen saturation. Treatment is usually divided into 2-3 daily treatments. Positioning should be used while using other treatments such as ACBT, percussions and vibrations.[4]
- While in position the individual can have their chest percussed for a few minutes. This can be followed or enhanced with deep breathing exercises, vibrations or huffing all working to cough up secretions.
- Percussions may be performed over the chest or back firmly and rhythmically usually over a layer of clothing or a towel so that it is comfortable and not painful in any way. These claps work to unstick thick mucus on the airways so that they can be mobilized to the central airways to come up in a cough.
- Vibrations are a technique that consists of several short rhythmical squeezes and shakes to the chest while the individual exhales in order to mobilize secretions.[4]
Modified postural drainage can be used to avoid gasto-oesophageal reflux. Studies show that a modified physiotherapy regimen without head-down tilt in infants with cystic fibrosis was associated with fewer respiratory complications than infants who are treated with a head tilt. [8]
Positive Expiratory Pressure[edit | edit source]

Positive Expiratory Pressure (PEP) is a technique that encourages breathing against resistance, which can be performed either through a device or against pursed lips.[20] The increase in pressure is transmitted to airways creating back pressure stenting them during exhalation, therefore preventing premature airway closure.[21] Not only does PEP prolong expiratory flow which prevents the airways it can also aid movement of mucus to the larger airways for easier expectoration.[21] There are many devices that can aid PEP:
- PEP Mask - the mask works to increase lung volume by mobilizing, transporting and evacuating secretions. By breathing out with a medium force through a resistance provided by the valve, the positive pressure allows airflow to get beneath the areas of mucus obstruction and move the mucus towards the larger airways where it can be coughed out.[13]
- Acapella Device - this device combines oscillating therapy and positive expiratory pressure therapy. Oscillating therapy are airway vibrations that help to shake or vibrate secretions off the walls of the airways. These vibrations are made by a lever with a magnet on the end inside the acapella. When air is blown out of the longs through the acapella valve, the airflow moves the lever back and forth creating vibrations to move into the airways. Please click on the video below to watch the acapella device in use.
- Flutter Device - Is a pocket device that is used to improve ventilation and mucus production by providing positive expiratory pressure and oscillations from a steel ball that vibrates under a sealed cover. Oscillation in this device can be changed depending on the angle that its held in. This device is great for individuals who are able to do therapy alone. The person must be compliant, responsible and able to control the angle of the device according to the oscillations they can feel in their lungs. The fultter device is difficult to use with young children and manual chest physiotherapy might be more effective at these stages.[9]
Evidence for PEP[edit | edit source]
- Short term studies with CF patients have shown the flutter to be similar to postural drainage and percussion or PEP
- Compared with autogenic drainage over a 4 week study period, the flutter showed no differences in sputum weight or lung function but viscoelasticity was significantly reduced with the flutter
- Konstan et al. reported that up to 3 times more sputum was produced with the flutter than with postural drainage but again Pryor et al found that significantly more sputum was produced with the active cycle of breathing techniques than with the flutter but that similar sputum weights were found over a 24-hour period.
- Two studies found no difference in lung function or exercise tolerance in children with exacerbated CF in hospital who used the flutter compared to those who were treated with percussion, vibration and postural drainage
- One study over 1 year in children with CF compared the flutter with the positive expiratory pressure mask and found a greater decline in forced vital capacity, increased hospital admissions and increased antibiotic use with the flutter[13]
- Flutter valve therapy has been suggested as an acceptable alternative to standard CPT during in hospital care of patients with CF[9]
Exercise[edit | edit source]
It is important to understand that not all physical activity is considered exercise. Be aware of the difference between exercise and physical activity.[4]
- Physical activity may be defined as any muscular contraction that creates a movement of the human body that causes an increase in resting energy expenditure
- Exercise may be defined as an organised form of physical activity consisting of repetitive movements with the intent to either maintain or build one's fitness capacity
It is important to be aware of the benefits and risks involved in leisure-time physical activity. [8] Individuals with CF are particularly vulnerable with respect to their reduced pulmonary function [9][10]
- Improved mucus clearance and sputum expectoration
- Improved posture
- Reduce effort of breathing at rest
- Reduce rate of disease progression
- Maintain bone mineral health and reduce risk of developing osteoporosis
- Reduce risk of developing diabetes
- Improve exercise tolerance
- Improved quality of life
- Increased feeling of breathlessness during exercise
- Higher risk of injuries compared to the healthy population during participation in contact or dangerous sports activities
- Pneumothorax (<1%)
- Asthma Attack
- Irritation of pre-existing arthritis
- Lung haemorrhage
- Loss of consciousness
(Reported in <1% of people who exercised)[8]
In addition to the benefits outlined specifically for those with CF universal exercise benefits are applicable. For instance, exercise has been shown to generate positive effects on mood and lower anxiety. Research also tells us that good experiences of physical activity can promote self-efficacy thereby increasing one’s confidence [10][7]. Such experiences have psychological benefits that those living with cystic fibrosis may also enjoy and so enable them to have similar experiences to their healthy peers.
Exercise Prescription[edit | edit source]
Recently, there has been a significant amount of research regarding the use of exercise as “medicine” to benefit individuals living with cystic fibrosis.[10][13] The importance of these studies is to deduce the physical capacity at which individuals with CF are capable of enduring. Furthermore, an meta-analysis of the results allows for clinicians to determine appropriate guidelines specifically designed for individuals with cystic fibrosis [7]
Swisher et al. [7] developed specific exercise prescriptions for individuals of all ages who live with cystic fibrosis; these can be viewed in the table below. It should be noted that, although these guidelines were developed by exercise and medical professionals, the onus is on the individual and their supportive medical and social teams to design a suitable personal programme; the exercises should clearly fit the needs, capabilities, and safety of the person performing the activity.

Exercise is often prescribed as a combined treatment for CF along with traditional respiratory physiotherapy techniques because movements are able to affect the physiological function of the body. For example, the movement of the body during exercise may help to condition the muscles thereby helping an individual to maintain correct posture. Having the correct posture can prevent curvatures in the spine that can ultimately lead to compression of the torso, which affects the ability to expand the chest during inhalation. Schindel et al. [3] examined this idea in detail, with a gold standard study design, and their research suggests that maintaining posture through mobility, stretching, and aerobic exercise can positively affect lung function, particularly for those with cystic fibrosis. Furthermore, Pedersen and Saltin [13] explain that the movements produced by exercise can stimulate optimal function of the mucociliary transport system, within the lungs, to aid in easier clearance of excess mucus due to the disease. With less mucus in the lungs there may be less chance of chest infection therefore enabling a better quality of normal breathing to be experienced. Such improvements in ventilation would allow the body to naturally increase the amount of air available for gaseous exchange in the lungs, thereby increasing oxygen into the blood, and ultimately successfully amending the individual’s oxygen saturation measures. Aside from exercise helping functions directly related to the pulmonary system it may also be used to decrease the chance of acquiring co-morbidities such as osteoporosis and diabetes.
Medical Management[edit | edit source]

Lung disease associated with Cystic Fibrosis (CF) is the cyclical pattern of mucous retention, infection, inflammation, and tissue damage. There are multiple therapies that address each one of the stages in that pattern.[4] Below are some of the emerging therapies available for CF patients.
- Mucous Clearance Therapies - While there are a number of physical techniques for mucous clearance, the emerging drug therapy research provides the CF community with pharmacological advances that create thinner mucous, and hydrate the airway surfaces to encourage easier mucous removal.[4]
- Airway Surface Rehydration Therapies
- Hypertonic Saline - Contribution to CF airway disease can be associated with increased sodium absorption from the airway surface liquid and the ensuing decrease in airway surface liquid volume. Inhalation of hypertonic saline can be used to counteract the airway surface liquid decrease by drawing water into the airway lumen. This type of therapy has shown to improve clearance of mucous, overall lung function, and reduction in pulmonary exacerbation frequency in patient with CF.[4]
- Mannitol - Dry-powder Mannitol (Marketed as Bronchitol, Pharmaxis) is an alternative therapy to hypertonic saline.[4] As a non-absorbable sugar alcohol, Mannitol aids in mucous clearance by rehydrating the airway surface and increasing the volume of airway surface liquid.[8] In a Phase III clinical trial, a sustained increase in FEV1 and decrease in pulmonary exacerbations were found, however, adverse effects including cough and coughing with blood occurred. From current studies, Mannitol has proven safe for CF patients that are able to tolerate the therapy. While the therapy has not been clearly proven to be effective in children, a Phase II clinical trial is underway.[8]
- P-1037 - CFTR interacts with proteins, including the epithelial sodium channel (ENaC) and due to normal inhibitory function loss; there is over-stimulation of ENaC and hyper-absorption of sodium, which leads to the dehydration of the airway surface and mucous. Currently, there is a Phase II clinical trial progression for P-1037; an inhaled ENaC blocker. Hyperkalaemia has been noted as a potential side effect of P-1037 due to renal exposure, and therefore, a low dose is required.[8]
- Anti-Infective Therapies - In Cystic Fibrosis, antibiotics are utilized under four conditions: prophylactically, infection eradication, chronic infection suppression, and exacerbation treatment.[8] CF pathogens found in the lungs vary with age, where S. aureus is the most common in infancy, H. influenzae increasing in childhood, and P. aeruginosa being the most common pathogen by adolescence and young adulthood.[8]
- Prophylaxis - UK and European guidelines currently recommend anti-staphylococcal antibiotics starting at diagnosis until approximately 3 years old as it has proven to reduce methicillin-susceptible S. aureus (MSSA) incidence, however, clinical outcome improvements have not been confirmed.[8] In contrast to this, USA recommendations are currently against the use of prophylactic anti-staphylococcal antibiotics due to results in one trial that reported increased pseudomonas infection rates (Edmondson and Davies, 2016). Further clinical trials of quality need to be completed to resolve the contrast between these recommendations for prophylactic therapy (Edmondson and Davies, 2016).
- Early Infection Eradication - In terms of infection, P. aeruginosa is the organism of most concern, due to the high risk it will become a chronic infection if not treated aggressively. Lung function decline can be associated with the inflammatory reaction of a chronic infection.. Early eradication programs are comprised of +/- systemic antibiotics, but can vary between countries. Inhaled tobramycin is used initially in North America, however, a multicentre trial in Europe is analyzing whether IV or oral antibiotics administered with nebulized colistin are of higher quality.[8]
- Chronic Infection Suppression The treatment emphasis changes from infection eradication to infection suppression once it becomes a chronic condition, and the intention is to decrease inflammation. Current research has concentrated on antimicrobial delivery through inhalation, which allows for high concentrations of the drug to be transported to the infection site, all while decreasing adverse effects and optimizing bacterial elimination. However, some adverse effects have been recognized, such as possible bronchospasm, displeasing taste, and length of time required to administer the drug. Currently, tobramycin, colistin, and aztreonam are nebulized antibiotics used to treat P. aeruginosa, and are often used in an alternating approach.[8] P
- Exacerbation Treatment - Pulmonary exacerbations are groupings of symptoms and decline in lung function and depending on the severity, are treated using oral or IV antibiotics. Patients will often receive a 2-week course of IV antibiotic, but research from one study has shown that maximal lung function is achieved following 10 days, and there is no advantage to continuing treatment past the 10 day mark. The CF Foundation in the USA has focused largely on this subject in a multicentre research programme.[8]
- Non-antibiotic Treatment Options to Bacterial Infection
- OligoG - In CF airways, chronic P. aeruginosa grows in a biofilm, which consists of airway mucins, exopolysaccharide, and a matrix of neutrophil DNA, and is largely resistant to antibiotic treatment.[8] OligoG is a sea-weed-derived alginate oligosaccharide formulated into a dry-powder agent, which obtains both anti-mucolytic and anti-biofilm characteristics and is currently in phase II trials.[8]
- IgY Antibodies - In a small clinical trial, it is suggested that protection against P. aeruginosa could be gained via IgY acquired from immunized hen eggs, and there is a multicentre trial underway which investigates the IgY antibody delivered as a gargle solution.[8]
- Anti-Inflammatory Therapies
- Non-steroidal Anti-inflammatory Agents - Ibuprofen has shown some improvement in younger patients with milder CF, however, current research has shown difficulties with dosing, where low-dose ibuprofen can encourage inflammation, and high-dose ibuprofen causes adverse side effects. In a Cochrane review completed recently, ibuprofen in higher doses showed slower lung function decline and decreased hospital stay, but long-term adverse effects were not investigated.[8]
- Therapies Targeting the CFTR Defect - Research targeting the CFTR defect focuses on three different groups of drug therapy which include potentiators, correctors, and read-through agents. With potentiator drug therapy, the CFTR channel activity is enhanced so long as it is located correctly.. Corrector drug therapy is used to repair defects that may include misfolding of the F508del protein, which allows cell surface trafficking. Lastly, read-through agents produce a full length protein by permitting a ribosome to dismiss a premature termination codon. In current research, these three types of drug therapies have either advanced to or through clinical trials.[8]
- Potentiators - Cystic Fibrosis treatment progress in more recent years has gained strength due to the development of ivacaftor, where the most common CF gene mutation (Asp551Gly (G551D)) has seen positive results in clinical trial with this agent. Improvements were seen in exacerbation rate, quality of life, weight, and FEV1 (~10% absolute improvement) during the ivacaftor trial, which has led to a license for use in CF patients aged 6 years and older. Further to this, a clinical trial in children ages 2-5 found similar results in sweat chloride; the CFTR biomarker.[8]
- Correctors - In a 2012 study, Lumacaftor (VX-809) was found to restore CFTR function to roughly 15% of CFTR level in vitro, however, this did not show significant improvements in F508del patients. Further to this, single-agent ivacaftor did not show significant changes, which was hypothesized to be a consequence of limited CFTR available at the cell surface.
- Read-through Agents - Production of a full length CFTR can be a result of Ataluren, which promotes ribosomal read-through of premature termination codons. In a phase III trial, there were no significant results seen between the ataluren and placebo groups, however, notable benefits were seen in patients who received aminoglycoside antibiotics through inhalation.
- CFTR Gene Therapy - There have been over 20 clinical trials in gene therapy since the CFTR gene was discovered, however many of these trials have focused on molecular or electrophysiological defects and single dose.[8] Recently, the UK CF Gene Therapy Consortium (GTC) has coordinated a phase IIb clinical trial of liposomal CFTR gene therapy where patients were administered nebulized doses, every month, for one year, or a placebo. Results showed that patients receiving the intervention had FEV1 stabilization, and there was a 3.7% statistical difference between intervention and placebo groups and further trials will investigate whether higher or more frequent doses can make additional improvements.[8] There is another approach to gene therapy, where the aim is to correct opposed to replace the gene, and a phase Ib clinical trial of nebulized mRNA repair molecule QR010 is currently in progress.[8]

Transition from Paediatric to Adult Care[edit | edit source]

Cystic fibrosis is no longer a condition the only affects children. Individuals with cystic fibrosis have a greater lifespan, and there are currently more adults with cystic fibrosis than children. There is a requirement to react to the developing maturity of adolescents with cystic fibrosis. They must be involved in decisions regarding their care and treatment. This should begin as soon as possible in the paedriatric service and continue within the adult service. Adolescents may not realise the need for transfer to begin with but it will become relevant to them as they mature, gain independence, interests and develop new relationships.
They will have to deal with managing their health and treatment along with education, career and relationships in the future. Therefore, it is necessary that they start to understand how their feelings and attitude about their health and treatment will infuence their future well-being.[8]
Differences Between Services[edit | edit source]
The paediatric service team work mainly with the parents rather than the child with CF. The parents are shown how to care for their child and manage their treatment. The childs decisions are made for them rather than with them. However, this gradually changes as the child develops and they will activily engage in making decisions with regard to their care.[9]
The adult service works directly with the individual with CF. The individuals should now be involved in the planning and delivery of health services under the guidance of the CF team.[10][7][13] The team aims to incorporate the treatment with the patient’s lifestyle and give emotional support to the individual as issues arise.
Parents and carers may still remain involved, however their role becomes supportive once the individual has transferred.
Timing[edit | edit source]

The discussion of transition should begin at least a year before the transfer to adult services occurs. This provides the individual and their parent/caregiver with sufficient time to consider feelings and settle any worries either may have.[8] The timing of transfer is different for individuals and is dependent on their requirements. However, to prevent premature or late transfer, it is advised that transfer should occur between the ages of 14 and 18.[3]
Barriers[edit | edit source]
Transition to adult care for young people with CF can be ineffective when there is:
- A lack of support of the transition process by any of the participants, which include the health care teams, adolescents and families, and the health care system;
- Limited preparation for transition;
- Lack of regular communication between participants.[5]
Effective Transitioning[edit | edit source]
Preparation, planning and communication by every participant (healthcare team, family and the individual) are key for a successful transition. This could be through ensuring the individual has been educated about CF and has the skills training to manage their long-term condition. If not, it is important to contact the relevant CF team member to address any concerns. It has been shown that young people with CF experience an effective transition when these is an are the opportunity for a joint meeting with the paediatric and adult services, a visit to the adult service before transition and having their first appointment made for them. If this is not the case, this should be highlighted with the CF team to ensure optimal transition.[6]
Role of Physiotherapists[edit | edit source]
What are Physiotherapists’ roles in transition?
- To communicate with the adult service about each individual’s specific treatment program to ensure effective transition.
- To promote self-management (hyperlink to “what is self-management?’ below) and give information about the adult service to the individual.
- To remain in contact with the adult service for the first year after transfer to adult services to ensure optimum healthcare.
- To listen to what the patient has to say and support them and their family.
- To respect the individuals decision whatever it may be.[4]
References[edit | edit source]
- ↑ 1.0 1.1 The Frey Life.What Is Cystic Fibrosis?Available from:https://www.youtube.com/watch?v=llrxGuU5o5c
- ↑ 2.0 2.1 2.2 Moran A. Cystic Fibrosis-Related Diabetes. Pediatric Endocrinology: Mechanisms, Manifestations, and Management. 2004:467
- ↑ 3.00 3.01 3.02 3.03 3.04 3.05 3.06 3.07 3.08 3.09 3.10 3.11 Anguiano A, Oates RD, Amos JA, Dean M, Gerrard B, Stewart C, Maher TA, White MB, Milunsky A. Congenital bilateral absence of the vas deferens: a primarily genital form of cystic fibrosis. Jama. 1992 Apr 1;267(13):1794-7.
- ↑ 4.00 4.01 4.02 4.03 4.04 4.05 4.06 4.07 4.08 4.09 4.10 4.11 4.12 4.13 4.14 4.15 4.16 4.17 4.18 4.19 4.20 4.21 Quittner, A., Abbott, J., Georgiopoulos, A., Goldbeck, L., Smith, B., Hempsted, S., Marshall, B., Sabadosa,K., & Elborn, S. International Committee on Mental Health in Cystic fibrosis: Cystic fibrosis Foundation and European Cystic fibrosis Society consensus statements for screening and treating depression and anxiety. British Medical Journal 2016; 71:26-34.
- ↑ 5.0 5.1 5.2 5.3 Di Sant'Agnese PA, Davis PB. Cystic fibrosis in adults: 75 cases and a review of 232 cases in the literature. The American journal of medicine. 1979 Jan 31;66(1):121-32.
- ↑ 6.0 6.1 6.2 6.3 6.4 Berschneider HM, Knowles MR, Azizkhan RG, Boucher RC, Tobey NA, Orlando RC, Powell DW. Altered intestinal chloride transport in cystic fibrosis. The FASEB Journal. 1988 Jul 1;2(10):2625-9.
- ↑ 7.0 7.1 7.2 7.3 7.4 7.5 7.6 7.7 7.8 Donaldson SH, Boucher RC. Pathophysiology of cystic fibrosis. Annales Nestlé (English ed.). 2007 Feb 22;64(3):101-9.
- ↑ 8.00 8.01 8.02 8.03 8.04 8.05 8.06 8.07 8.08 8.09 8.10 8.11 8.12 8.13 8.14 8.15 8.16 8.17 8.18 8.19 8.20 8.21 8.22 8.23 8.24 8.25 Zielenski J. Genotype and phenotype in cystic fibrosis. Respiration. 2000 Apr 7;67(2):117-33.
- ↑ 9.0 9.1 9.2 9.3 9.4 9.5 9.6 Gibson RL, Burns JL, Ramsey BW. Pathophysiology and management of pulmonary infections in cystic fibrosis. American journal of respiratory and critical care medicine. 2003 Oct 15;168(8):918-51.
- ↑ 10.00 10.01 10.02 10.03 10.04 10.05 10.06 10.07 10.08 10.09 10.10 10.11 8Wilschanski M, Durie PR. Patterns of GI disease in adulthood associated with mutations in the CFTR gene. Gut. 2007 Aug 1;56(8):1153-63.
- ↑ Cystic fibrosis and transition. Cystic Fibrosis Trust. Accessed from https://www.cysticfibrosis.org.uk/what-is-cystic-fibrosis/cystic-fibrosis-care/transition
- ↑ Emotional Wellness. Cystic Fibrosis Foundation. Accessed from https://www.cff.org/Life-With-CF/Daily-Life/Emotional-Wellness/
- ↑ 13.00 13.01 13.02 13.03 13.04 13.05 13.06 13.07 13.08 13.09 13.10 13.11 13.12 13.13 Kaplan E, Shwachman H, Perlmutter AD, Rule A, Khaw KT, Holsclaw DS. Reproductive failure in males with cystic fibrosis. New England Journal of Medicine. 1968 Jul 11;279(2):65-9.
- ↑ Cystic Fibrosis Foundation. Anxiety and CF. Accessed from https://www.cff.org/Life-With-CF/Daily-Life/Emotional-Wellness/Anxiety-and-CF/
- ↑ Cystic Fibrosis Foundation Airway Clearance Techniques (ACTs) Accessed fromhttps://www.youtube.com/watch?time_continue=238&v=1Ufj3oU_M2w
- ↑ Cat Young Autogenic Drainage Available fromhttps://vimeo.com/107231066
- ↑ Kevin Viken Postural Drainage Available from https://www.youtube.com/watch?time_continue=14&v=ywIkbcf_HOA
- ↑ KEBMS10 Postural Drainage Positions Available fromhttps://www.youtube.com/watch?time_continue=9&v=6EfGqTpFAjc
- ↑ kclontzbell.Cystic Fibrosis Physical Therapy of Toddler part 2. Available fromhttps://www.youtube.com/watch?v=hDo3RvGyPgs
- ↑ Fagevik Olsén M, Lannefors L, Westerdahl E. Positive expiratory pressure e Common clinical applications and physiological effects.Respiratory Medicine.2015;105(3):297- 307.
- ↑ 21.0 21.1 Darbee JC, Ohtake PJ, Grant BJ, Cerny FJ. Physiologic evidence for the efficacy of positive expiratory pressure as an airway clearance technique in patients with cystic fibrosis. Phys Ther. 2004;84(6):524-37

