Wilson's Disease
Original Editors - Students from Bellarmine University's Pathophysiology of Complex Patient Problems project.
Top Contributors - Rachel Rawson, Lexi Vessels, Aminat Abolade, Kim Jackson, Vidya Acharya, WikiSysop and Elaine Lonnemann
Definition/Description[edit | edit source]
Wilson’s disease, also known as hepatolenticular degeneration, is an autosomal recessive disorder that results from abnormal metabolism of copper.[1] It is caused from the mutation of the ATP7B gene, and leads to the accumulation of copper in key organs such as the liver, central nervous system, kidney, cornea and other tissues, resulting in impaired function.[2] The deposition of copper begins immediately at birth, with symptoms usually presenting in late adolescence. Wilson's disease typically manifests into two common presentations: Liver disease or neurological symptoms, due to these organ's large involvement in the accumulation of copper. Symptomatic adolescents will tend to experience more symptoms related to liver pathologies, while older adults will experience neurological signs and symptoms.[3]
Prevalence[edit | edit source]
Wilson’s disease is a rare condition, affecting only one person in 30,000 in most populations.[4] This condition is considered an autosomal recessive. The gene frequency for this disease has been found to be 56%, with a carrier frequency of 1 in every 90 people. According to recent studies, an increase incidence of this gene mutation has been found in isolated populations like Costa Rica and Sardinia, an island off Italy.[3]

Characteristics/Clinical Presentation[edit | edit source]
- The main involvements are most commonly hepatic, neurological, and psychiatric and these symptoms can present from asymptomatic to fatal if left untreated.[3]
- Hepatic presentation can present as asymptomatic, acute hepatitis, acute/chronic liver failure, and cirrhosis. Other symptoms that may present are, Coomb's negative hemolytic anemia, low grade hemolysis and jaundice.[3]
- Neurological involvement can present with athetoid movements and unsteady gait. They can also present similar to Parkinson's, ataxia, pseudosclerosis dominated by tremor, and a dystonic syndrome that often leads to contractures.[3]
- Other central nervous system symptoms include pill rolling tremors in hands, facial and muscular rigidity, and dysarthria.[2]
- Psychiatric characteristics can present in the hand-eye coordination, emotional and behavioral changes, and decrease in performance.[3]
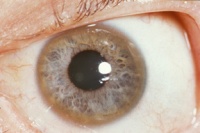

- Wilson's can also have an ocular manifestation as a Kayser-Fleischer ring around the eye fromcopper deposits[2] and a sunflower cataracts in the lens, which is less commonlyobserved compared to the Kayser-Fleischer ring.[3]
- Liver disease is most likely the manifestation in the pediatric population and neuro disease is most common in young adults.[2]
Associated Co-morbidities[edit | edit source]
- Individuals with Wilson's disease can present with a variety of different complications that are directly related to the excess of copper in targeted organs; however, in severe cases other systems can become affected as well. There can be musculoskeletal manifestations including muscle atrophy and wasting, contractures, deformities, osteomalacia, and pathological fractures.[2]
- Other less common characteristics that can present because of Wilson's disease are osteoarthritis, abnormal kidney function tests, and cardiomyopathy.[1]
Medications[edit | edit source]
Pharmacological treatment of Wilson’s disease is based around the removal of excess copper from the body, as well preventing the accumulation of it in target organs. Chelation therapy drugs, such as Penicillamine and Trientine are used to remove excess copper from the affected organs; they act as binding agents and are removed through urinary excretion. Zinc salts are also used to block the intestinal absorption of copper, reducing its accumulation in the body. Zinc salts are shown to be very effective, and is an advantage over other pharmacological agents because of its lack of side effects.[4]
It is recommended that patients who are asymptomatic should receive lower doses of zinc salts or chelators than patients who are symptomatic. Often times, symptomatic patients receive a combination of zinc salts and chelators.[4]
Medicines:
D-Penicillamine:
Initial Dose: 250-500 mg/d with 250 mg increments every 4-7 days in 2-4 divided doses Maintenance Dose: 750-1000 mg/d in 2 doses a day Pediatric Dose: 20mg/kg per day in 2-3 doses daily; maintenance therapy should reduce to 25-30% of dose
Administer drug one hour before or two hours after a meal to avoid inhibition.
Adverse side effects are common and include fever, cutaneous eruptions, neutropenia, lymphadenopathy, and proteinuria[3]
Trientine Dosage:
Initial Dose: 750-1500 mg/d in 2-3 divided doses Maintenance Dose: 750-1000 mg/d in 2-3 divided doses Pediatric Dose: 20mg/kg per day in 2-3 doses daily; maintenance therapy should reduce to 25-30% of dose
Adverse side effects include possible neurotoxicity, dyspepsia, anemia, muscle cramps, and dystonia[3]
Zinc Dosage: Dose: 150 mg/d divided in 3 doses Pediatric dose: 75 mg/d divided in 3 doses
Adverse side effects are minimum and include gastric irritation, alcohol intolerance, headaches, excessive perspiration, and anemia.[3]
Compliance with pharmacological intervention poses a significant threat to management of Wilson's disease, especially in patients who are asymptomatic because there is failure to recognize the relationship between medication and the disease progression.[4]
Diagnostic Tests/Lab Tests/Lab Values[edit | edit source]
Diagnosis of Wilson’s disease can be confirmed through a multitude of diagnostic tests:
Ophthalmologic slit-lamp examination for Kayser-Fleischer rings[4]
Serum ceruloplasmin test: levels <2mg/dL will be seen in individuals with Wilson's disease.[4]
24-hour urine copper test: Levels of copper in urine are increased in WD because there is a decrease in serum binding of copper to ceruloplasmin. Levels in an adult > 100mcg/hr and levels in children > 40mcg/hr confirm WD.[4]
Liver biopsy for histology and histochemistry and copper quantification. This is the gold standard for determining the presence of Wilson's disease.[1]
Genetic testing, haplotype analysis for siblings and mutation analysis[4][3]
Due to the heterogeneity of the clinical manifestations, misdiagnosis is common and this leaves patients without timely treatment.[1]
Aetiology/Causes[edit | edit source]
Wilson's Disease is a disorder that results in excess copper accumulation in target organs and impairing the normal function of the particular organs (liver, brain, cornea, etc). The exact cause of this disease is not fully understood, however, the gene that is currently believed to be responsible is the mutation of ATP7B. This gene, ATP7B, is a vitally important enzyme for copper transport in the body. The mutation in this gene is linked to abnormal excretion and ultimately deposition of copper in particular organs. It is believed that other factors may be involved with the development of Wilson's, such as other genetic variations, lipid metabolism, and possible environmental factors. Further research on the aetiology of Wilson's Disease is needed.[1]
Systemic Involvement[edit | edit source]

- Hepatic: Jaundice, abdominal swelling (ascites), vomiting blood, abdominal pain, oesophageal varices, asymptomatic hepatomegaly, Isolated splenomegaly, Persistently elevated serum aminotransferase activity (AST, ALT), fatty liver, acute hepatitis, resembling autoimmune hepatitis, cirrhosis: compensated or decompensated, and acute liver failure.[4]
- Neurological: Tremors, unsteady gait, ataxia, dysphagia, dysarthria, dystonia, dyscoordination, pseudobulbar palsy, dysautonomia, migraine headaches, insomnia, and seizures.[4]
- Psychiatric: Mental illness including homicidal or suicidal behavior, depression, aggression, and insomnia[4]
- Other Systems:
Ocular: Kayser-Fleischer rings, sunflower cataracts
Cutaneous: lunulae ceruleae
Renal abnormalities: aminoaciduria and nephrolithiasis
Skeletal abnormalities: premature osteoporosis and arthritis
Cardiomyopathy, dysrhythmias
Pancreatitis
Hypoparathyroidism
Menstrual irregularities; infertility, repeated miscarriage[4]
Medical Management (current best evidence)[edit | edit source]
Wilson’s disease is a rare condition, but with proper management the progression of the disease can stop and even in some cases, symptoms can improve. Immediate treatment early on in the disease progression is crucial in order to prevent irreversible damage to the liver and other organs.[4]
The most important aspect of medical management for Wilson’s disease is compliance to pharmacological intervention. The goals of treatment are based on the removal of excess copper from the body through the use of chelator drugs, and blocking the intestinal absorption of copper through the use of zinc salts.[3]
Medications:
D-penicillamine (Cuprimine and Depen) was the first drug introduced to remove excess copper from the body through increased urine excretion. While it is commonly used amongst these patients, a disadvantage to this medication is the adverse side effects it brings with it, toxicity and worsening neurological symptoms.[3]
Trientene (Syprine and Trientine Dihydrochloride) is an alternative for patients who are intolerant to penicillamine. This drug acts in a similar manner, but without the serious side effects.[3]
Zinc (Galzin) acts by inhibiting the absorption of copper in the intestinal tract. An advantage to this drug over chelators is the effectiveness and lack of adverse side effects.[3]
Treatment for Wilson’s disease is lifelong, and adherence to therapy provides a challenge for a considerable amount of patients, especially asymptomatic patients. Patient education on medication compliance is extremely important in this population. Managing liver disease is an important aspect of the medical management of Wilson’s disease, and can lead to failure if left untreated. Patients with serious hepatitis or liver failure will not improve with pharmacological intervention, and may require a liver transplant.[3]
Additional therapy is necessary for treatment of this condition. Specifically, physical therapy can be beneficial to improve neurological symptoms, such as dystonia, dyscoordination, and difficulty with balance. Speech therapy can improve other symptoms like dysarthria and dysphagia.
Diet is a crucial component in medical management. Patients should stick to a diet that is low in copper and avoid foods like certain meats, mushrooms, chocolate, shellfish, and nuts. Water should be tested for any copper content to avoid excessive copper intake.[4]
Physical Therapy Management (current best evidence)[edit | edit source]
Physical therapy can play a major role in the treatment of individuals with Wilson's disease. One of the most common manifestations of this condition is the neurological symptoms that present like a movement disorder. Patients present with dystonia, dyscoordination, tremors and difficulty with balance and walking.[4]
Many patients may face problems if their dystonia worsens and a contracture develops. PT can provide patient education on positioning and stretching to prevent progression of the contracture, in addition to serial casting to reverse an acute contracture.[4]
Physical therapy can be a useful tool in improving balance and coordination through the use of therapeutic activities and exercise, as well as maintaining range of motion.[4][5]
Patients who suffer from liver disease due to the effects of Wilson's disease may experience fatigue, muscle and joint pain, and a decrease in functional status. Physical therapy can play a role in decreasing pain and improving the patient's functional capabilities with energy conservation in mind.[6]
While physical therapy plays a crucial role in helping these individuals overcome their neurological symptoms, success with therapy is not possible unless the patient is consistently sticking to their medication regime.[3]
Differential Diagnosis[edit | edit source]
- Cirrhosis
- Haemochromatosis
- Portal Hypertension
- Hemorrhage associated with oesophageal Varices
- Hematochromatosis
- Essential Tremor
Case Reports/ Case Studies[edit | edit source]
Malik A, Khawaja A, Sheikh L. Wilson’s disease in pregnancy: case series and review of literature. BMC Research Notes [serial on the Internet]. (2013, Nov), [cited April 7, 2016]; 6(1): 1-8. Available from: Academic Search Complete.
Grover S, Sarkar S, Jhanda S, Chawla Y. Psychosis in an adolescent with Wilson's disease: A case report and review of the literature. Indian Journal Of Psychiatry [serial on the Internet]. (2014, Oct), [cited April 7, 2016]; 56(4): 395-398. Available from: Academic Search Complete.
Loudianos G, Incollu S, Mameli E, Lepori MB. Wilson’s disease in an adult asymptomatic patient: a potential role for modifying factors of copper metabolism. Annals of Gastroenterology : Quarterly Publication of the Hellenic Society of Gastroenterology [serial on the Internet]. (2016, Jan-March), [cited April 7, 2016]; 29(1):96-98.
Raju K, Bangalore G, Thuruvekere S, Pathavanalli V. Wilson's disease: A Clinical autopsy case report with review of literature. Journal Of Natural Science, Biology & Medicine [serial on the Internet]. (2015, Jan), [cited April 7, 2016]; 6(1): 248-252. Available from: Academic Search Complete.
Resources[edit | edit source]
Wilson Disease Association (WDA) International
U.S. National Library of Medicine
References[edit | edit source]
- ↑ 1.0 1.1 1.2 1.3 1.4 Fei W, Jing W, Chunwen P, Liang Q, Chunmeng J. Wilson's Disease: A Comprehensive Review of the Molecular Mechanisms. International Journal Of Molecular Sciences [serial on the Internet]. (2015, Mar), [cited April 8, 2016]; 16(3): 6419-6431. Available from: Academic Search Complete.
- ↑ 2.0 2.1 2.2 2.3 2.4 Goodman CC, Fuller KS. Goodman and Fuller’s Pathology E-Book: Implications for the Physical Therapist. Elsevier Health Sciences; 2020 Oct 9.
- ↑ 3.00 3.01 3.02 3.03 3.04 3.05 3.06 3.07 3.08 3.09 3.10 3.11 3.12 3.13 3.14 3.15 3.16 Rodriguez-Castro K, Hevia-Urrutia F, Sturniolo G. Wilson's disease: A review of what we have learned. World Journal Of Hepatology [serial on the Internet]. (2015, Dec 18), [cited April 8, 2016]; 7(29): 2859-2870. Available from: MEDLINE.
- ↑ 4.00 4.01 4.02 4.03 4.04 4.05 4.06 4.07 4.08 4.09 4.10 4.11 4.12 4.13 4.14 4.15 4.16 Wilson Disease Association (WDA) International [Internet]. Wilson Disease Association (WDA) International. [cited 2016Mar30]. Retrieved from: http://www.wilsonsdisease.org/
- ↑ Brewer G, Askari F. Wilson's disease: clinical management and therapy. Journal Of Hepatology [serial on the Internet]. (2005), [cited April 8, 2016]; 42 Suppl(1): S13-S21. Available from: MEDLINE.
- ↑ van den Berg-Emons RJ, van Ginneken BT, Nooijen CF, Metselaar HJ, Tilanus HW, Kazemier G, Stam HJ. Fatigue after liver transplantation: effects of a rehabilitation program including exercise training and physical activity counseling. Physical Therapy. 2014 Jun 1;94(6):857-65.
- ↑ Goodman CC, Snyder TK. Differential Diagnosis for Physical Therapists: Screening for Referral. 5th Edition. St. Louis, Missouri: Elsevier Saunders, 2013.

