Cystic Fibrosis
Original Editor - Rachael Lowe and The Open Physio project. Top Contributors - Erica Essex, Admin, Jake Warren, Kim Jackson, Rachael Lowe, Laura Ritchie, Uchechukwu Chukwuemeka, Vidya Acharya, Kristin Hamrick, 127.0.0.1, Mande Jooste, Lucinda hampton, WikiSysop, Shaimaa Eldib, Meaghan Rieke, Evan Thomas, Naomi O'Reilly and Michelle Lee
Clinically Relevant Anatomy [edit | edit source]
Cystic Fibrosis is an inherited disorder that can lead to a variety of clinical manifestations. In an attempt to better understand the pathology of this disorder it is important to analyze the specific anatomical structures that will be impacted by this disease process.[1]

- Bronchioles: Obstructed secondary to the thickened mucus adhering to the walls of the airways.
- Small intestine: distal obstruction (meconium ileus)
- Pancreatic duct: Obstruction
- Bile ducts: Obstruction
- Male reproductive organs: absent or obstructed vas deferens
- Female reproductive organs: cervical canal obstructed by thickened mucus
- Musculoskeletal system:
- Inspiratory muscle atrophy
- Weakness/atrophy in anti- gravity muscles such as the gastrocnemius
- Kyphosis of the spine resulting in neck and back pain
Mechanism of Injury / Pathological Process[edit | edit source]

Cystic fibrosis (CF) is the most frequent cause of suppurative lung disease in the younger Caucasian population. A depleted volume of the airway surface liquid (ASL) layer in the respiratory system leads to abnormal mucociliary clearance[2]. A chronic cycle of infection and inflammation results in progressive suppurative bronchiectasis and lung damage.
Cystic Fibrosis is an inherited disease of the mucus and sweat glands (exocrine glands) affecting mostly the lungs, liver, pancreas and intestines[3][4]. It causes damage to lung tissue, inflammation, and acute susceptibility to bacterial infections. There is an abnormal gene, called Cystic Fibrosis Transmembrane Conductance Regulator (CFTR), which results in the production of thick, sticky mucus which blocks the airways in the lungs resulting in frequent lung infection[4]. The thick mucus also blocks the ducts in the pancreas which in turn blocks digestive pancreatic enzymes from reaching the small intestine and performing their normal function.
The degree of mutation of the CFTR gene determines the type of complications that will arise and these will differ from person to person. The mutated or defected protein attaches to the outer membrane of cells in sweat glands, lungs, pancreas and other affected organs[4], and spans over the membrane which acts as a channel connecting the inner part of the cell cytoplasm to the surrounding fluid. This channel is important in our airways because it controls the movement of chloride from the inside to the outside of the cell. Chloride moves from the sweat into the cytoplasm but due to the mutation of the CFTR protein, the chloride is trapped inside the cells of the airway and outside the skin. Chloride is a negatively charged ion and sodium is a positively charged ion, so this leads to an electrical attraction, which results in the formation of salt. In CF patients a high amount of salt is lost in sweat, thus forming the basis of the sweat test.[5]
There are 3 theories:
- The lack of chloride exodus through the CFTR protein leads to the accumulation and an increase in the viscosity of the nutrient-rich mucus in the lungs leading to bacteria hiding from the body’s immune system.
- There is a paradoxical increase in the sodium and chloride uptake due to the CFTR protein defect. This cause an increase in the water reabsorption which leads to thick and dehydrated mucus.
- This theory focuses on abnormal chloride movement out of the cell which leads to dehydrated mucus, pancreas, secretion, biliary secretion etc
These theories propose that the major damage in patients with CF is due to the blockage of narrow passages of the affected organs due to thick secretions. The blockage within the lung airways leads to infections because of the accumulation of the enzymes and blockages of the passages.
Prevalence or Incidence Around the World[edit | edit source]
In the United States, the prevalence is 1 case per 3200 people for white people, for black people the prevalence is 1 case per 15 000 people, in hispanics the prevalence is 1 case per 9 200 people and in Asian Americans the prevalence is 1 case per 31 000 people. (Girish & Sharma, 2008). Another study showed that approximately 30 000 children and adults in the US and 70 000 individuals worldwide suffers from CF (Grief, 2008). CF is fatal, but with improvement of medical research and treatment techniques the survival rate has increased. More than 40% of CF individuals are 18years or older.
It is estimated that one in every 2,500 babies born in the UK will be born with cystic fibrosis and there are more than 9,000 people living with the condition. [6].
In Canada, approximately one in every 3,600 children born has cystic fibrosis [7].
Survival rate for CF patients from the UK born in 1978: 55% of males and 49% of females by 2003 (25 years)
Epidemiology in the UK: 2003 mid-year CF population:[8]
| 0–<1 | 1– <5 | 5– <10 | 10– <15 | 15 | 1 | 17-<25 | 25-<35 | 35– <45 | 45–55 | >55 | Total | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Male | 140 | 550 | 700 | 725 | 152 | 143 | 992 | 672 | 318 | 40 | 7 | 4439 |
| Female | 140 | 550 | 700 | 634 | 128 | 140 | 802 | 479 | 221 | 39 | 12 | 3845 |
| Total | 280 | 1100 | 1400 | 1359 | 280 | 283 | 1794 | 1151 | 539 | 79 | 19 | 8284 |
| % | 3.4 | 13.3 | 16.9 | 16.4 | 3.4 | 3.4 | 21.7 | 13.9 | 6.5 | 1.0 | 0.2 | 100.00 |
Causes and Risk Factors[edit | edit source]
Cystic Fibrosis is an autosomal recessive disease caused by defects in the CFTR gene[9]. This CFTR gene normally encodes for a protein that functions as a chloride channel and is regulated by cyclic AMP, but in cystic fibrosis, the CFTR gene mutates and results in abnormalities of cyclic AMP, which regulates the transport of chloride across the epithelial cells on mucosal surfaces. This defective gene causes the body to produce an abnormally thick and sticky fluid in the respiratory and GI tracts, the pancreas and in the sweat glands.
In order to develop CF, two CF genes must be inherited, one from the mother and the other from the father. If only one CF gene is inherited, then the person is called a carrier and will have no symptoms. When both parents are carriers, with each pregnancy there is a: 1 in 4 chance of having a child with cystic fibrosis, 1 in 2 chance of having a child who is a carrier or 1 in 4 chance of having an unaffected child.
There are several different types of genetic mutation which are associated with different degrees of severity of the disease.[10]
Signs and Symptoms[edit | edit source]

Cystic Fibrosis can be asymptomatic; this occurs when a person only inherits one defective gene from either of their parents and is a carrier. The pattern of development of CF and the severity of its symptoms varies among individuals. The disease is sometimes obvious soon after birth, but in some cases of CF, they are not detected for months (in infancy) or years (in childhood).
Signs and symptoms will differ from person to person and these may include:
- Fatigue
- Salty-tasting skin
- Persistent cough with phlegm
- Wheezing and shortness of breath
- Lung infections
- Poor growth and weight loss
- Difficulty with bowel movements in the first 24/48 hours of life.
Organs affected[edit | edit source]
CF is a "multi-system" disease, meaning that it affects many body organs. However, most of the symptoms involve the lungs and the digestive system.
Lungs[edit | edit source]
In a healthy person, there is a constant flow of mucus over the surfaces of the air passages in the lungs. This removes debris and bacteria. If you have CF, this mucus is excessively thick and sticky and cannot perform this role properly. The sticky mucus also provides an ideal environment for bacterial growth. This can put a person with CF at risk of getting bacterial chest infections and pneumonia. If these infections are not treated early and properly, they can cause severe complications. "Pulmonary involvement is the most common and severe manifestation of CF" (Goodman, 2013). Cystic Fibrosis begins as an obstructive lung disease in which patients have difficulty pushing the air out of their lungs, this results in hyperinflation of the lungs and bronchiectasis. If these lungs problems become chronic overtime it leads to further destruction of the lung tissue and fibrosis, leading to a restrictive lung disease on top of the already present obstructive lung disease. [11]
Pancreas[edit | edit source]
CF also affects the digestive system. In a healthy person, the pancreas produces chemicals (enzymes) which pass into the gut as food leaves the stomach. These enzymes break down the fat. In CF, the pancreas does not produce these enzymes and without them, the fat in food is not properly digested and it is difficult to gain weight. The faeces will contain an excess of fat and will be oily and have a putrid smell.
Intestines[edit | edit source]
Newborns may present with intestinal obstruction. Infants present with an increase in the frequency of stools and failure to thrive as a result of malabsorption. This intestinal obstruction is referred to as meconium ileus, in which the infants will have thick, puttylike, tenacious meconium. This is commonly the earliest manifestation of CF, but it is also important to note that all children with CF are susceptible to intestinal obstruction that commonly presents as impacted stools. [1]
Respiratory Tract[edit | edit source]
Patients present with a dry cough, which can be recurrent or chronic, and can also produce sputum. The patient may suffer from shortness of breath and chest pain. There may also be recurrent wheezing and pneumonia, pneumothorax and haemoptysis.
More specifically: In babies and infants, the symptoms of CF are:
- Persistent diarrhea
- Bulky, foul smelling and greasy stools
- Pale stools
- Frequent wheezing or pneumonia
- Chronic cough with thick mucus
- Salty-tasting skin
- Poor growth
- Blockage of the intestine (called meconium ileus)
- Abdominal swelling
- Gassiness
- Vomiting
- Dehydration
In children, the symptoms include:
- Frequent respiratory infections
- Fever
- Cough
- Difficulty in breathing
- Abdominal pain and discomfort
- Flatulence
- Fast respiration
- Flaring of the nostrils
- Poor appetite
- Malnutrition
- Poor growth
- A barrel-chested appearance
Genitourinary System[edit | edit source]
Infertility in men and women is a common clinical manifestation of CF. Men have decreased fertility secondary to the absence or obstruction of the vas deferens and women have decreased fertility secondary to the thickened mucus in the cervical canal. [1]
Musculoskeletal System[edit | edit source]
It is important for Physical Therapist to be aware of the clinical manifestations in the musculoskeletal system. Physical Therapists should be aware that these patients are likely to have decreased bone mineral density, which is going impact the spine. These patients will often have increased Thoracic Hyperkyphosis secondary to becoming barrel chested. Other manifestations may include muscle atrophy and myalgia. Adults who have been dealing with Cystic Fibrosis long-term may present with osteoarthropathy, Rheumatoid arthritis, and/or osteoporosis. [1]
Other medical problems may arise as a result of CF, such as:
- Sinusitis (inflammation of the nasal sinuses)
- Nasal polyps (fleshy growths inside the nose)
- Clubbing (rounding and enlargement of fingers and toes)
- Pneumothorax (rupture of lung tissue and trapping of air between the lung and chest wall)
- Coughing up blood
- Enlargement of the right side of the heart (called cor pulmonale)
- Protrusion of the rectum through the anus (called rectal prolapse)
- Liver, pancreatic and gallbladder problems
- Delayed puberty
Diagnosis[edit | edit source]
With a complete medical history and physical examination, diagnostic procedure may include the following:
- Sweat (chloride) Test: A test, which measures the amount of chloride in the sweat. If the amount of chloride is higher than the normal amounts it may suggest cystic fibrosis. The sweat test is not painful.
- Blood test: Blood or cheek scraping cells can also be used to detect mutations of the CFTR gene.
- Chest X-Rays: A diagnostic test, which produce images of internal tissues, bones and organs.
- Pulmonary Function Test: Measures the lungs' ability to exchange oxygen and carbondioxide appropriately.
- Sputum cultures: Is often performed to determine if an infection is present.
- Stool Evaluations: To measure stool fat absorption.
- Pancreatic function
- Computed Tomography(CT) Scan: Detect early pulmonary disease before symptoms occur.
- Magnetic Resonance Imaging(MRI): Is not only used to screen the lung, but also the heart, sinuses and GI tract.
- Prognosis
Although symptoms vary, consensus is that prognosis mainly depends on the extent of pulmonary involvement.[12] Deterioration usually occurs leading to debilitation and subsequent death due to respiratory failure and/or cor pulmonale. Prognosis has improved over the past fifty years because of advancement in the management, especially before the irreversible pulmonary changes. The median age for survival varies globally. A study conducted by Stephenson et al in 2017 reported that the median age of survival is increasing but there are still major differences between countries, with the median age of survival in Canada being 50.9 years, almost 10 years than in the USA 40.6 years[13]. There are several factors which account for long term survival rates in people with CF; good nutrition and patients without pancreatic insufficiency has better survival rates[14]. The following have been associated with worse prognosis; females, early colonization with mucoid Pseudomonas, presentation with respiratory symptoms, tobacco smoke exposure and airway hyperreactivity.
Lung function test especially FEV1, is the best predictor of survival depending on the age and sex of the patient[15].
Management / Interventions[edit | edit source]
Although airway clearance continues to be an integral part of care, physical exercise, postural care and the need to address the unique complications which emerge as a result of improved longevity have significantly changed the nature of physiotherapy in CF.
There is no cure for CF and most people with the disease die between the ages of 20 and 30 as a result of lung failure. Early diagnosis and a complete treatment plan can improve both survival and quality of life for the person suffering from CF.
Medical Management[edit | edit source]
Medical treatment of CF includes:
- Antibiotics for infections of the respiratory system
- Vitamin supplements
- Bronchodilators: Inhaled medicines to help open the airways
- DNAse enzyme replacement therapy to thin the mucus and makes it easier to expectorate
- Pain relievers
Enzyme therapy is done with each meal or snack, where most people with CF will need to take replacement enzymes such as pancreatin (eg Pancrex). These supply the missing pancreatic enzymes and allow proper digestion.[16] People with CF normally need vitamin and mineral supplements too.
The usual childhood vaccinations, such as measles, mumps and rubella (MMR) and diphtheria, tetanus and whooping cough (DTP) are important for the children with CF. If you have CF you should also be vaccinated against flu and pneumococcus to help prevent chest infections[17].
CF is one of the most extensively researched genetic diseases as a target for gene therapy development and this can also serve as an important model for gene therapy of other diseases. The aim of gene therapy is to correct the basic defect in CF. This can be achieved by the development of a vector. This can be done by doing pre-clinical and clinical research. Clinical studies have identified the host immune response and low vector efficiency as key impediments to effective CF gene therapy.[18] The study[19] provides recommendations to the Cystic Fibrosis (CF) community on the management of both common and unique issues that arise when individuals reach a state of Advanced cystic fibrosis lung disease (ACFLD).
Physiotherapy Management[edit | edit source]
The main aim of physiotherapy is to prevent secondary complications and improve quality of life by removing excessive mucus secretions, maintain or improve lung function and assist with musculoskeletal therapy where needed. (Rowe, 2009).
Chest Physiotherapy[edit | edit source]
A person suffering from CF will require intensive chest physiotherapy. This will include the following:
- Vigorous massage to help loosen the sticky mucus
- Postural drainage: gravity assisted positions to help drainage of secretions and also helps to increase the air movement or ventilation to different parts of the lungs [20].
- Percussion: This technique is also known as chest clapping, and is used to help loosen secretions. To perform this technique a cupped hand is used to clap the chest firmly and rhythmically (over a layer of clothing or a towel) [20].
- Shaking and Vibrations: This technique consists of several short rhythmical squeezes to the chest while exhaling to mobilize secretions [20].
- Regular assessment and monitoring is necessary during physiotherapy treatment as the patient may require supplemental oxygen, especially in advanced cystic fibrosis.
| [21] |
Other techniques include:
- Positioning: lying on either side, front or back and sitting [20]
- Active cycle of breathing (ACBT): This technique consist of Breathing Control (BC), Thoracic Expansion Exercises (TEE), and Forced Expiration Technique (FET)
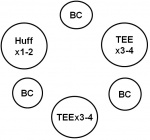

- Breathing Control (BC): Relaxed diaphragmatic breathing which allows for rest and helps avoid any tightening of airways, which can make it difficult to clear secretions [20]
- Thoracic expansion exercises (TEE): Deep breathing exercises that help the lungs to expand more effectively and allow air to get behind secretions so that they can be "pushed" up the airways towards the mouth. The breaths should be slow and deep with a pause at the end of inspiration then followed by relaxed quiet expiration [20].
- Forced Expiration Technique (FET): This consists of huffing or a sigh, to help move secretions from the smaller to the larger airways from where they can be cleared more easily[20].
- Positive expiratory pressure (PEP): A technique that helps to open up airways and get air behind secretions to help move them higher up the airway. The PEP device (mask or mouthpiece) gives a small degree of resistance to the breath out and this resistance splints open the airways. Bubble PEP is used in young children and involves the child blowing through a straw placed in a bottle of water and soap to produce bubbles.☃☃☃☃ A descriptive study at specialized treatment centers in Brazil assessed the respiratory therapy techniques for the treatment of patients with CF. It suggested high-frequency oral oscillation (HFO), huffing, and positive expiratory pressure (PEP) was frequently recommended[22].
- Oscillating PEP: These devices combine vibrations of airways with PEP and there are several different types. The Flutter is a small pipe shaped device that interrupts the flow of air and gives an intermittent back pressure to the airways as well as causing them to vibrate. The Cornet and the Acapella are two other oscillating PEP devices that function similarly to the Flutter device [20]
- Gentle huffing
- Minimize coughing
- Encourage aerobic exercises: participating in physically active activities or do exercises such as running, swimming, dancing etc
Parents of a child with CF are taught by hospital staff on how to manage their child. Older children and adults can be taught to do this for themselves. Advice will include all of the techniques above. It is also important to educate the family about the condition and to provide counselling where necessary.
| [23] |
Airway Clearance[edit | edit source]
Despite several comparative studies of airway clearance techniques in CF, there continues to be a lack of robust evidence to support the long term benefits of intervention[24]. A lack of evidence should however not necessarily be associated with a lack of benefit. Clinicians generally agree that airway clearance is likely to be beneficial in those with established respiratory disease[25].
Systematic reviews comparing the various airway clearance modalities have not identified a single technique as being superior to others[26][27]. Some studies report a trend for patients to prefer therapies which are self administered[28][29]. The performance of routine daily airway clearance require considerable time and can impose a significant burden on patients and families. Adherence to routine airway clearance continues to be reported as poor[30][31]. The use of non-invasive ventilation as an adjunct to airway clearance may be useful in improving gas exchange during sleep in those with moderate to severe disease but its impact on disease progression remains unclear[32].
The primary aim of airway clearance is to prevent or delay the onset of bronchiectasis and lung damage. In infants, modified postural drainage and percussion remain widely used internationally but other techniques such as infant PEP and assisted AD along with physical activity have emerged as feasible alternatives. Newborn screening (NBS) for cystic fibrosis, now widely available internationally, has led to much earlier diagnosis and the emergence of a cohort of healthy infants who appear free of clinical symptoms and the role of routine airway clearance in “asymptomatic infants has recently been questioned[33]. There is however strong evidence for the presence of very early lung disease in terms of inflammation and infection reduction in lung function and structural changes even in “pre-symptomatic” infants. This issue continues to generate much debate internationally and opinions remain divided[34]. Consensus methodology has been used by the Association of Chartered Physiotherapists in Cystic Fibrosis in the UK to develop guidelines for the physiotherapy management of asymptomatic infants with CF[35]. A change in the traditional approach should not be interpreted as a withdrawal of physiotherapy from babies who are apparently well but as a new approach to intervention.
Postural Drainage[edit | edit source]
| [36] |
The state-of-art review[37] suggests avoiding repeated airway clearance in infants and children with acute pulmonary disease.The association of gastro-oesophageal reflux and postural drainage using a head down tipped position, particularly in babies with CF has led to a significant change in practice internationally. Many centers now advocate only the use of modified Parkinson's (omitting a head down tip). The place of Parkinson's as a routine therapy has also been questioned in the absence of copious secretions, a situation no longer uncommon in the CF population[34].
A randomised crossover trial suggests positioning in side-lying during nebulisation improves the apical deposition of the inhaled drug in healthy adults and adults with mild cystic fibrosis lung disease[38].
Exercise[edit | edit source]
The short and long-term benefits of regular physical activity are well established and the importance of regular physical exercise as an essential part of the physiotherapy regimen is recognized. The effect of inspiratory muscle training in CF, however, remains unclear.st
Benefits of Exercise for the CF patient include (Goodman, 2009):
- Enhanced fitness levels
- Increased sputum clearance
- Delay the onset of dyspneoa
- Delay declines in pulmonary function
- Prevent decrease in bone density
- Enhance cellular immune response
- Improve quality of life
- May improve self-image, confidence and have psychological benefits.
Exercise in the Literature[edit | edit source]
- Recent literature suggests that exercise produces similar benefits as the traditional airway clearance techniques and exercise should be encouraged over these techniques when clinically appropriate for the patient. Exercise is beneficial in that evidence suggests that exercise may even reverse some of the disease related changes in ion transport resulting in a decrease of the thickened mucus and therefore leading to improved pulmonary functioning. [39]
- In a clinical trial by Elbasan et al. clinically stable children with cystic fibrosis (FEV1<35%) were taken through a training program that consisted of active cycles of breathing techniques and aerobic exercise training on the treadmill using 74-80% of the maximum heart rate for 30 minutes. This was performed three times a week for six weeks. The active cycles of breathing techniques were taught along with posture and breathing to be used as a home exercise program. The study concluded that active cycles of breathing techniques should be included with aerobic exercise to improve physical fitness including thoracic mobility, muscle strength and endurance, flexibility, and speed.[40]
- A systematic review of randomized clinical trials by Nancy van Doorn included four out of 599 RCT’s that met the inclusion criteria. Exercise interventions included aerobic training, resistance training, anaerobic training and strength training. Strength training was strongly correlated with strength gains which is important to prevent de-conditioning in this patient population. Results also suggested that peak VO2 could be improved from short-term aerobic training, which is important for survival rate in these patients. Anaerobic fitness also demonstrated improvement. This systematic review concludes that children with CF can improve pulmonary function through either aerobic or resistance training. [41]
- A recent trial study by Reix et al. sought to answer the questions: "1. Can session of exercise with incorporated maneuvers substitute for a session of breathing techniques for airway clearance in children with cystic fibrosis? and 2. Are children with cystic fibrosis as co-operative and satisfied with the exercise regimen as with the breathing techniques?" Using a randomized, crossover design "each participant underwent two 20 minute airway clearance interventions on two scheduled clinic days: One involving 3 bouts of various whole-body exercise modalities each followed by independent expiratory maneuvers , and the other involving breathing control, thoracic expansions with manual expiratory compression, and the forced expiratory technique." Although the conclusions of this study are very limited by design, they concluded that "a session of various whole-body exercises interspersed with independent expiratory maneuvers could be an acceptable substitute for breathing techniques" and that it may be preferred by children with cystic fibrosis due to the ability to modify the exercise bouts in order to keep it new and challenging.[42]
- A study by Paranjape et al. looked at the effects of a home exercise program for children with cystic fibrosis hypothesizing that increasing their activity would lead to improved exercise capacity, lung function, nutritional status, and quality of life. Their outcome measures included the Modified Shuttle Walk Test, BMI, FEV1, Habitual Activity Estimation Scale, and 5 exercise-related domains of the Revised CF Quality of Life Questionnaire. Measures were taken before and after the intervention which included a 2-month, individualized home exercise program. All measurements showed an increase or improvement except for the HAES on weekday activity but only the MSWT and CFQ-R body image perception domain were statistically significant. The conclusion of this study is that individualized, home exercise programs are beneficial for children with cystic fibrosis.[43]
- The study by Hoffman L et showed positive outcomes with inspiratory muscle training in patients with advanced lung disease[44]
- A recent randomised control study suggests people with Cystic Fibrosis well-tolerated Low-volume high-intensity interval training (HIIT), and low-volume high-intensity interval training (HIIT) showed positive effects on exercise capacity n physical function in people with CF.[45]
End-of-life care[edit | edit source]
The patient and the family need more sensitive discussions about prognosis and preferences for care throughout the course of the disease. Most people facing end-of-life with cystic fibrosis are older adolescents or young adults and should be informed of the choices they have about their illnesses. Often discussions about transplantation are done to give the patients the chance to weigh the merits of longer survival with a transplant against the deteriorating illness without a transplant.[46] Palliative care such as sedation should be given to ensure peaceful dying, when it is appropriate. Patients at this stage should consider trying the most aggressive treatment trials available within a limited time and to accept death when the trials did not work successfully.[47]
Differential Diagnosis [48][edit | edit source]
| Disease/Condition | Differentiating Signs/Symptoms | Differentiating Tests |
| Primary ciliary dyskinesia | Usually not associated with pancreatic insufficiency; chronic purulent middle ear infections, which are less common in children with CF. | Ciliary biopsy will demonstrate ultrastructural abnormalities of respiratory cilia, or genetic testing may reveal abnormalities in genes that encode key ciliary proteins. |
| Primary immunodeficiency | Severe combination of immune deficiency with respiratory infections, IgA deficiency, and IgG1 deficiency; may be associated with nonrespiratory infections. Usually not associated with pancreatic insufficiency. | Measurement of lymphocyte number and function, neutrophil function, and immunoglobulin levels. |
| Asthma | Usually not associated with purulent bronchitis. Patients with asthma usually will not demonstrate digital clubbing. | There is no diagnostic test. Diagnosis is made clinically. Some children with CF also present with asthma. |
| Gastroesophageal reflux disease (GERD) | Usually not associated with signs or symptoms of malabsorption. | Modified barium swallow or gastric emptying studies may be useful in making the diagnosis of chronic aspiration or GERD, but may also be positive in patients with CF. |
| Chronic aspiration | Usually not associated with signs or symptoms of malabsorption. | Modified barium swallow or gastric emptying studies may be useful in making the diagnosis of chronic aspiration or GERD, but may also be positive in patients with CF. |
| Failure to thrive | May not be associated with respiratory symptoms. | Sweat test should be negative if CF is not the cause. However, severe malnutrition causes a falsely elevated sweat test. |
| Celiac disease | Patients with celiac disease will respond to removal of gluten from the diet. | Intestinal biopsies. |
| Protein losing enteropathy | Loss of serum protein through the GI tract. Can be associated with Fontan procedure, lymphatic disorders, or mucosal erosion. | Intestinal biopsies. |
link: https://online.epocrates.com/u/2935403/Cystic+fibrosis
Outcome Measures[edit | edit source]
Cystic Fibrosis Questionnaire-Revised (CFQ-R)
Cystic Fibrosis Questionnaire (CFQ)
Baseline and Transition Dyspnea Indexes (BDI-TDI)
The next 7 outcome measures were found in a review of Clinically useful outcome measures for physiotherapy airway clearance techniques[49]
1. FVC
2. FEV1
3. Ratio between FEV1 and FVC
4. Blood gas analysis (Pulse oximetry)
5. Sputum quantity
6. Auscultation
7. Computer Aided lung sounds analysis
Pulmonary Function: measured using FVC and FEV1 percentages
Aerobic Capacity: measured using VO2 (ml/kg/min) changes. One study suggests that a regular peak VO2 exercise test should be performed regularly in these patients to monitor function over time. Additionally this study suggests that peak VO2 is a good predictor of mortality and that patient's response to exercise is related predictor of morbidity. [39]
Key Evidence[edit | edit source]
Annual Evidence Update on Cystic Fibrosis – Physiotherapy, S Ammani Prasad, Cystic Fibrosis Unit, Great Ormond Street Hospital for Children NHS Trust, for NHS Evidence, 2009.
Resources[edit | edit source]
Physiotherapy National Standards of Care for people with Cystic Fibrosis, Association of Chartered Physiotherapists in Cystic Fibrosis, UK, 2009.
Association of Chartered Physiotherapists in Cystic Fibrosis, this website can be used for personal information purposes, copyrighted by CSP, 2014.
Physiotherapy works: This is a valuable patient resource that includes a pdf and subsections that include: What is Cystic Fibrosis (CF)?, What is the role of physiotherapy?, Prevention of airway damage caused by increased resistance and obstruction, Exercise is very important in the management of CF, Conclusion, and a Case Study. This information is available for personal use through the Association of Chartered Physiotherapists, Copyright 2014.
Case Studies[edit | edit source]
Cystic Fibrosis Case Study example from a textbook The case study is on page 3 of the pdf document, it provides a case example of an infant who was diagnosed with CF and how the CF developed and impacted him as he aged.
References[edit | edit source]
- ↑ 1.0 1.1 1.2 1.3 Goodman CC, Fuller KS. Pathology: Implications for the Physical Therapist. 3rd ed. St.Louis: Saunders Elsevier; 2009. p788 – 799.
- ↑ Mall M, Boucher RC. Pathogenesis of pulmonary disease in cystic fibrosis. In: Bush A, Alton EWF, Davies JC, Griesenback U, Jaffe A (Eds) Cystic Fibrosis in the 21st century Progress in Respiratory Research Karger, Basel 2006; 34:116-121
- ↑ O'Sullivan BP, Freedman SD. Cystic fibrosis. Lancet. 2009;373 (9678): 1891–904
- ↑ 4.0 4.1 4.2 Sheppard MN, Nicholson AG.The pathology of cystic fibrosis. Current Diagnostic Pathology. 2002; 8(1):50–59
- ↑ Quinton PM. Cystic fibrosis: lessons from the sweat gland. Physiology. 2007;22(3):212-25.
- ↑ Nhs.uk. Cystic fibrosis - NHS Choices, 2015. Available from: http://www.nhs.uk/conditions/cystic-fibrosis/Pages/Introduction.aspx [Accessed 4th June 2015].
- ↑ Cysticfibrosis.ca. What is Cystic Fibrosis? - Cystic Fibrosis Canada. Available from: http://www.cysticfibrosis.ca/about-cf/what-is-cystic-fibrosis/ [Accessed 4th June 2015].
- ↑ Dodge J, Lewis P, Stanton M, Wilsher J. Cystic fibrosis mortality and survival in the UK: 1947-2003. European Respiratory Journal. 2007;29(3):522-526.
- ↑ Sharma G. Cystic Fibrosis. Medscape. Available from: https://emedicine.medscape.com/article/1001602-overview. (accessed 15 August 2019).
- ↑ Hoffman LR, Ramsey BW. Cystic fibrosis therapeutics: the road ahead. Chest. 2013;143(1):207-13.
- ↑ Goodman C, Snyder T. Differential Diagnosis for Physical Therapist Screening for Referral. 5th ed. St. Louis: Elsevier Saunders; 2013. p307.
- ↑ Naehrig S, Chao CM, Naehrlich L. Cystic Fibrosis: Diagnosis and Treatment. Deutsches Ärzteblatt International. 2017;114(33-34):564.
- ↑ Stephenson AL, Sykes J, Stanojevic S, Quon BS, Marshall BC, Petren K, et al. Survival comparison of patients with cystic fibrosis in Canada and the United States: a population-based cohort study. Ann Intern Med. 2017; 166:536–547
- ↑ Singh VK, Schwarzenberg SJ. Pancreatic insufficiency in Cystic Fibrosis. Journal of Cystic Fibrosis. 2017; 16:S70–S78.
- ↑ Taylor-Robinson D, Whitehead M, Diderichsen F, Olesen HV, Pressler T, Smyth RL, et al. Understanding the natural progression in% FEV1 decline in patients with cystic fibrosis: a longitudinal study. Thorax. 2012;67(10):860-6.
- ↑ Somaraju UR, Solis-Moya A. Pancreatic enzyme replacement therapy for people with cystic fibrosis. Cochrane Database Syst. Rev. 2014;(10):CD008227.
- ↑ Burgess L, Southern KW. Pneumococcal vaccines for children and adults with cystic fibrosis. Cochrane Database Syst Rev 2016;(9):CD008865
- ↑ Wagner JA, Gardner P. Toward cystic fibrosis gene therapy. Annu Rev Med. 1997;48:203-16.
- ↑ Kapnadak SG, Dimango E, Hadjiliadis D, Hempstead SE, Tallarico E, Pilewski JM, Faro A, Albright J, Benden C, Blair S, Dellon EP. Cystic Fibrosis Foundation consensus guidelines for the care of individuals with advanced cystic fibrosis lung disease. Journal of Cystic Fibrosis. 2020 Feb 27.
- ↑ 20.0 20.1 20.2 20.3 20.4 20.5 20.6 20.7 Prasad S, Orska T, Ferguson K, Agent P, Dodd M, Dhouibe E et al. Physiotherapy treatment in cystic fibrosis: airway clearance techniques [Internet]. 1st ed. Bromley: Cystic Fibrosis Trust; 2013 Available from: http://www.cysticfibrosis.org.uk/media/151218/FS%20-%20Physiotherapy_airway%20clearance_v4_Apr_2013.pdf ([accessed 26 May 2015).
- ↑ enmurrcp. RT Chest Physiotherapy Demonstration. Available from: http://www.youtube.com/watch?v=ErMTXJLE5es[last accessed 08/02/13]
- ↑ Donadio MV, Campos NE, Vendrusculo FM, Stofella AM, da Silva Almeida AC, Ziegler B, Schivinski CI, Santuzzi CH, Sarges ED, Gonçalves FM, de Oliveira Ribeiro MÂ. Respiratory physical therapy techniques recommended for patients with cystic fibrosis treated in specialized centers. Brazilian Journal of Physical Therapy. 2019 Nov 29.
- ↑ American Lung Association. Teach a child to belly breathe for relaxation. Available from: http://www.youtube.com/watch?v=VOnDA6_MAWI[last accessed 08/02/13]
- ↑ van der Schans CP, Prasad A, Main E. Chest physiotherapy compared to no chest physiotherapy for cystic fibrosis. Cochrane Database of Systematic Reviews 2000; (2): CD001401. DOI: 10.1002/14651858.CD001401
- ↑ Flume PA, Robinson K, O’Sullivan B, Finder JD, Vender FL, Willey-Courand DB, et al. Cystic Fibrosis Pulmonary Guidelines: Airway Clearance therapies. Resp Care 2009;54(4):522-537
- ↑ Main E, Prasad SA, van der Schans C. Conventional chest physiotherapy compared to other airway clearance techniques for cystic fibrosis. The Cochrane Database of Systematic Reviews 2005;(1): CD002011
- ↑ Elkins M, Jones A, van der Schans CP. Positive expiratory pressure physiotherapy for airway clearance in people with cystic fibrosis. Cochrane Database of Systematic Reviews 2006;(2): CD003147
- ↑ Morrison L, Agnew J. Oscillating devices for airway clearance in people with cystic fibrosis. Cochrane Database of Systematic Reviews 2009;(1): CD006842
- ↑ Pryor JA. A comparison of five airway clearance techniques in the treatment of people with cystic fibrosis. Thesis submitted for the degree of Doctor of Philosophy, Imperial College London 2005
- ↑ Bucks RS, Hawkins K, Skinner TC, Horn S, Seddon P, Horne R. Adherence to Treatment in Adolescents with Cystic Fibrosis: The Role of Illness Perceptions and Treatment Beliefs. J Pediatr Psychol. 2009;34(8):893-902
- ↑ Arias Llorente RP, Bousoño García C, Díaz Martín JJ. Treatment compliance in children and adults with cystic fibrosis. J Cyst Fibros. 2008;7(5):359-67
- ↑ Moran F, Bradley JM, Piper AJ. Non-invasive ventilation for cystic fibrosis. Cochrane Database Syst Rev 2007;(4):CD002769
- ↑ Pryor JA, Main E, Agent P, Bradley JM. Physiotherapy. In: Bush A, Alton E, Griesenbach U, Jaffe A (Eds). Cystic Fibrosis in the 21st century. vol 34. London:Prog Respir Res. Basel, Karger; 2006. p301-308
- ↑ 34.0 34.1 Prasad AS. Annual Evidence Update on Cystic Fibrosis – Physiotherapy, NHS Evidence, 2009. Available from:http://www.library.nhs.uk/respiratory/ViewResource.aspx?resID=311642 [Accessed 22nd August 2019)
- ↑ Association of Chartered Physiotherapists in Cystic Fibrosis, UK. Physiotherapy National Standards of Care for people with Cystic Fibrosis. 2009 Available from:http://cms.interactivecsp.org.uk/uploads/documents/ACPCF%20National%20Standards%20of%20Care..doc (accessed 8 March 2013)
- ↑ Lord of Phisiotherapy. Postural Drainage. Available from: http://www.youtube.com/watch?v=TPZsP1ujg0U[last accessed 08/02/13]
- ↑ Morrow BM. Airway clearance therapy in acute paediatric respiratory illness: A state-of-the-art review. South African Journal of Physiotherapy. 2019 Jun 25;75(1):12.
- ↑ Dentice RL, Elkins MR, Verschuer J, Eberl S, Dwyer G, Bye PT. Side lying during nebulisation can significantly improve apical deposition in healthy adults and adults with mild cystic fibrosis lung disease: a randomised crossover trial. BMC pulmonary medicine. 2019 Dec 1;19(1):128.
- ↑ 39.0 39.1 Cerny F. Exercise and Cystic Fibrosis (CF) 2.0. Pediatric Exercise Science. 2013;25(4):616-23.
- ↑ Elbasan B, Tunali N, Duzgun I, Ozcelik U. Effects of chest physiotherapy and aerobic exercise training on physical fitness in young children with cystic fibrosis. Italian Journal of Pediatrics. 2012; 38(2): 1-5.
- ↑ van Doorn N. Exercise programs for children with cystic fibrosis: a systematic review of randomized controlled trials. Disability and Rehabilitation. 2010; 32(1): 41-49.
- ↑ Reix P, Aubert F, Werck-Gallois MC, Toutain A, Mazzocchi C, Moreux N, et al. Exercise with incorporated expiratory maneuvers was as effective as breathing techniques for clearance with cystic fibrosis: a randomized crossover trial.J Physiother. 2012; 58(4): 241-47.
- ↑ Paranjape S,Barnes LA, Carson KA, von Berg K, Loosen H, Mogayzel Jr, PJ. Exercise improves lung function and habitual activity in children with cystic fibrosis. Journal of Cystic Fibrosis. 2012; 11(1); 18-23.
- ↑ Hoffman M, Augusto VM, Eduardo DS, Silveira BM, Lemos MD, Parreira VF. Inspiratory muscle training reduces dyspnea during activities of daily living and improves inspiratory muscle function and quality of life in patients with advanced lung disease. Physiotherapy theory and practice. 2019 Aug 21:1-1.
- ↑ Sawyer A, Cavalheri V, Jenkins S, Wood J, Cecins N, Bear N, Singh B, Gucciardi D, Hill K. High-Intensity Interval Training Is Effective at Increasing Exercise Endurance Capacity and Is Well Tolerated by Adults with Cystic Fibrosis. Journal of clinical medicine. 2020 Oct;9(10):3098.
- ↑ Sands D, Repetto T, Dupont LJ, Korzeniewska-Eksterowicz A, Catastini P, Madge S. End of life care for patients with cystic fibrosis. J Cyst Fibros. 2011;10(2):S37-44. doi: 10.1016/S1569-1993(11)60007-6.
- ↑ Beers MH, Sharp M, Dohme Research Laboratories (Rahway NJ.). The Merck manual of geriatrics. Whitehouse Station, NJ : Merck Research Laboratories, 2006.
- ↑ Epocrates. Cystic Fibrosis. Available from: https://online.epocrates.com/u/2935403/Cystic+fibrosis/Diagnosis/Differential (accessed 20 August 2019)
- ↑ Marques A, Bruton A, Barney A. Clinically useful outcome measures for physiotherapy airway clearance techniques: a review. Available from: http://www.academia.edu/441672/Clinically_useful_outcome_measures_for_physiotherapy_airway_clearance_techniques_a_review (accessed 20 August 2019)

